How to use
How to run the webtool
To run MHCcombine you need an HTML5 compatible browser, this means Internet Explorer 9, Firefox 4, Chrome 18 or higher versions.
How to use MHCcombine
1) Enter the sequence of your protein of interest in FASTA format or a list of PEPTIDES separated by a new line:
SEQUENCE(S)
Get the amino acid sequence of your protein of interest in FASTA format. E.g. copy it from UniProtKB database. Avoid copying of line breaks.You can also enter multiple oligopeptides, e.g. sequence stretches which contain a mutation-based amino acid change to predict neoepitopes.
Sequences will be splitted into peptides of the length selected in step 5.
FASTA example (Protein):
>sp|P03126|VE6_HPV16 Protein E6 OS=Human papillomavirus type 16 OX=333760 GN=E6 PE=1 SV=1
MHQKRTAMFQDPQERPRKLPQLCTELQTTIHDIILECVYCKQQLLRREVYDFAFRDLCIV
YRDGNPYAVCDKCLKFYSKISEYRHYCYSLYGTTLEQQYNKPLCDLLIRCINCQKPLCPE
EKQRHLDKKQRFHNIRGRWTGRCMSCCRSSRTRRETQL
FASTA example (Oligopeptides):
>peptide1
PEPTIDEPEPTIDEPEPTIDE
>peptide2
EPITQPEEPITQPEEPITQPE
>peptide3
SEQRENCESEQRENCESEQRENCE
PEPTIDE(S)
List the amino acid sequences of your peptides of interest seperated by line breaks. Predictions will be performed with the actual length of entered peptides.PEPTIDE LIST example:
PEPTIDEPEP
EPITQPEEPI
SEQRENCESE
2) Select the MHC species/loci of interest:
Choose between a list of representatives of HLA supertypes and lists of HLA-A, HLA-B or HLA-C alleles.
3) Select the MHC type of interest:
Choose the desired HLA-type from the list selected in step 2). Multiple HLA-types can be requested per query and each selected HLA-type will be added to the "Selected alleles" field below.
4) Select the prediction methods:
Choose all algorithms you want to apply by ticking the respective checkboxes or simply select all methods by activating "Select all".
5) Select the peptide length:
Choose the desired sequence length of the predicted peptides. You can either request each length separately or select 8-11 to predict 8-,9-,10- and 11-mers at the same time. Please note that not all methods are capable of predicting all peptide lengths.
6) Enter an ID for your protein sequence of interest:
For your convenience, you can enter up to 10 characters to identify the query. This could be e.g. the name of your protein. The ID will be used to create the output file name.
7) Submit your request
Clicking the submit button will start the queries. Activate the "Hide" option to remove unselected methods from the columns in the output table. Once all jobs finished you can save the output as a comma seperated value (.csv) file that is named according to your selected ID, peptide length and HLA-type. The download will be available for 30min.
8) For publication of results, please check How to cite.
Whenever using our tool to combine the output of prediction methods for publications please cite our tool and the used methods.
How to convert the output .csv-file
The .csv-file can be opened with every spreadsheet platform. Here we describe the use of Microsoft Excel.
Manually import the .csv-file to assure correct formatting.
1) Open a new blank Excel file and click:
Data->Get External Data->from Text...
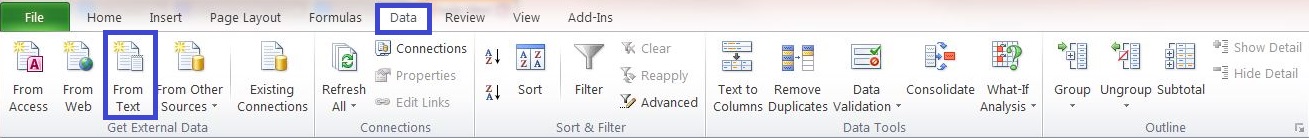
2) Select the .csv-file and use the Text Import Wizard.
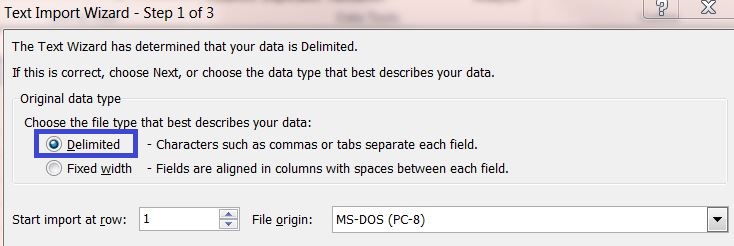
3) Select the original data type "Delimited" and continue to the next page.
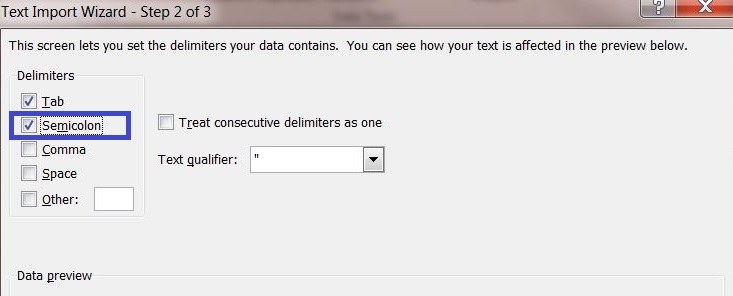
4) Select the delimiter "Semicolon" and continue to the next page.
5) In data preview select the columns "Region" and "Sequence" and change the column data format to Text.
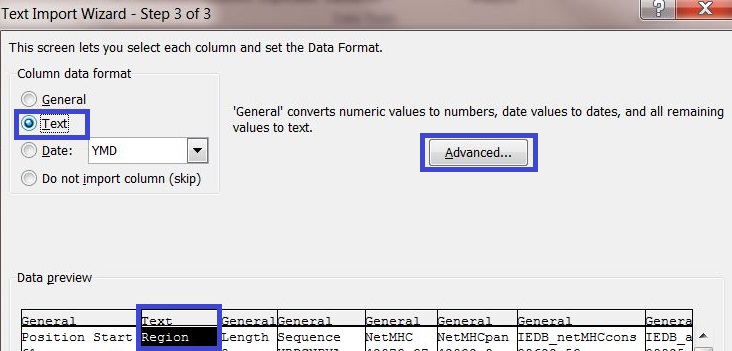
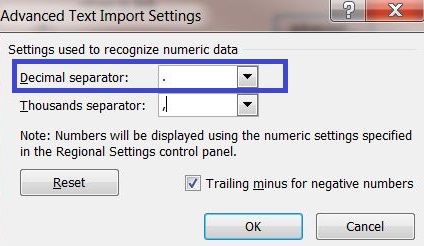
6) Click on the button "Advanced..." and enter a dot "." as decimal separator to adequately convert the server output into your system settings.
7) Select the A1 field and click:
Insert->Table...
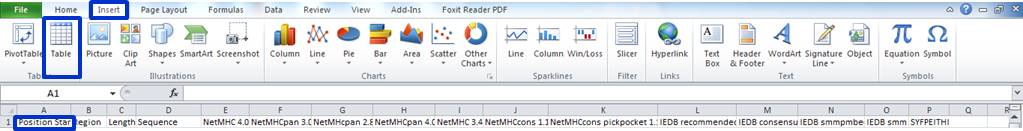
Select the whole range of output data to convert all rows and columns into a sortable table. Now it should look like this:
